imputation文献-A systematic evaluation of single-cell RNA-sequencing imputation methods
文章题目
A systematic evaluation of single-cell RNA-sequencing imputation methods
中文名:
单细胞RNA测序插补方法的系统评价
文章地址:
https://doi.org/10.1186/s13059-020-02132-x
本帖只探讨设计假阳性的检测部分,其余不做补充。
评价插补方法:

多种插补方法的评测:
评估了
(1)基于模型的插补方法(bayNorm[19]、SAVER[20]、SAVER-X[21]、scImpute[22]、scRecover[23]、VIPER[24])
(2)基于平滑的插补法(DrImpute[25]、MAGIC[26]、kNN平滑[27])
(3)使用深度学习方法(AutoImpute[28]、DCA[29]、DeepImpute[30]、SAUCIE[31]、scScope[32]、scVI[33])
(4)基于低秩矩阵的方法(ALRA[34]、mcImpute[3]、PBLR[36])的数据重建方法
比较思路:
- 通过比较同质细胞群中的插补细胞轮廓和批量样本之间的相似性,评估插补方法本身在恢复真实表达值方面的性能
- 研究了下游分析中插补方法的性能,包括差异表达分析、无监督聚类和轨迹分析
使用数据:
- 模拟数据
- 使用 plast-based和droplet-based的两种scRNA-seq数据
有关假阳性的测试部分:
1.DEG分析
假设:多次测试校正后 同质细胞不会出现DEG
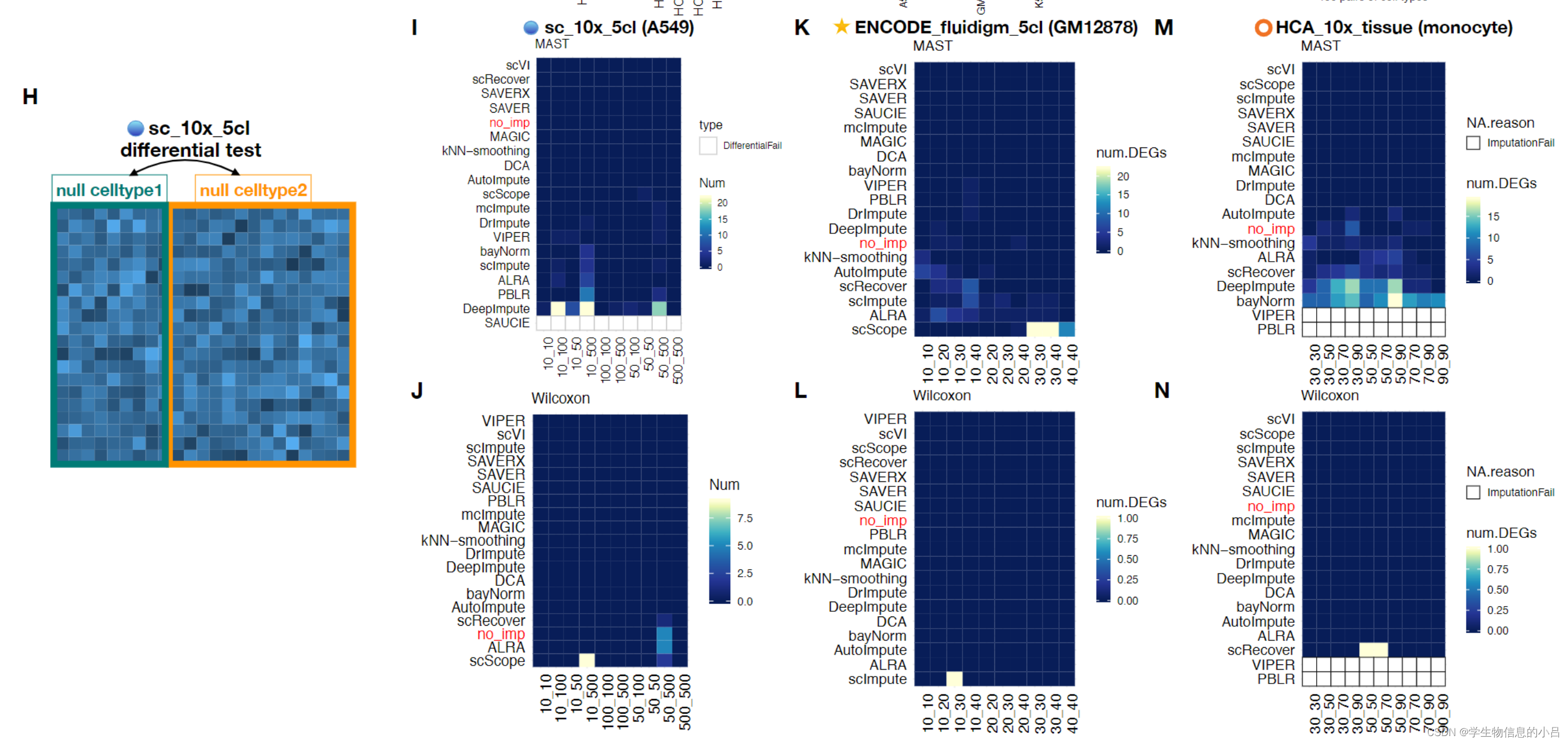
试验方法:
同一细胞类型分成两组细胞 X_Y X和Y分别代表每一组的细胞个数
在不做插补和做完各类插补后 两组之间的差异性 计算MAST和Wilcoxon两种index
结果:
不同类型的插补方法识别假阳性DEG的方法并不一致,因为所有类型的插拔方法(基于模型、基于平滑和基于数据重建)都有报告假阳性DEGs的例子。同时,当识别DEG时,细胞群大小不平衡导致更多的插补方法使用MAST和Wilcoxon报告假阳性
2.补充第一条
问题:已知细胞类型特异性标记基因的插补表达是否能够正确预测细胞类型(使用真-数据集)

数据集
使用基于UMI和FACS分类的外周血单个核细胞(PBMC)数据集(PBMC_10x_tissue)和在纯化的PBMC细胞类型中高度表达的已知PBMC标记基因(CD19用于B细胞;CD14用于单核细胞;CD34用于CD34+细胞;CD3D用于CD4 T辅助细胞、细胞毒性T细胞、记忆T细胞、细胞毒T细胞、调节性T细胞;CD8A用于细胞毒性T淋巴细胞和细胞毒T淋巴细胞)
试验方法:
评估了基于标记基因(如CD19)表达预测细胞类型(如B细胞)的性能。
估计了ROC(AUROC)曲线下的面积,其中标记基因的表达(如CD19表达)是预测因子,而真正的细胞类型(如B细胞或非B细胞)是标签
结果:
11种插补方法中有10种(返回插补数值的)产生的AUROC高于没有插补的方法
3.DE占数据集中基因的比率对插补方法的影响
已知
在识别DEG的背景下,DE对插补法的性能至关重要
方法
使用大量RNA序列数据将细胞系成对和分层基因分为高LFC和低LFC,其中高(或低)LFC基因定义为基于LFC绝对值的前10%(或后10%)基因。
LFC-Log Fold change:具体含义看下面这篇帖子:
DEG计算指标-LFC和P-value
结果
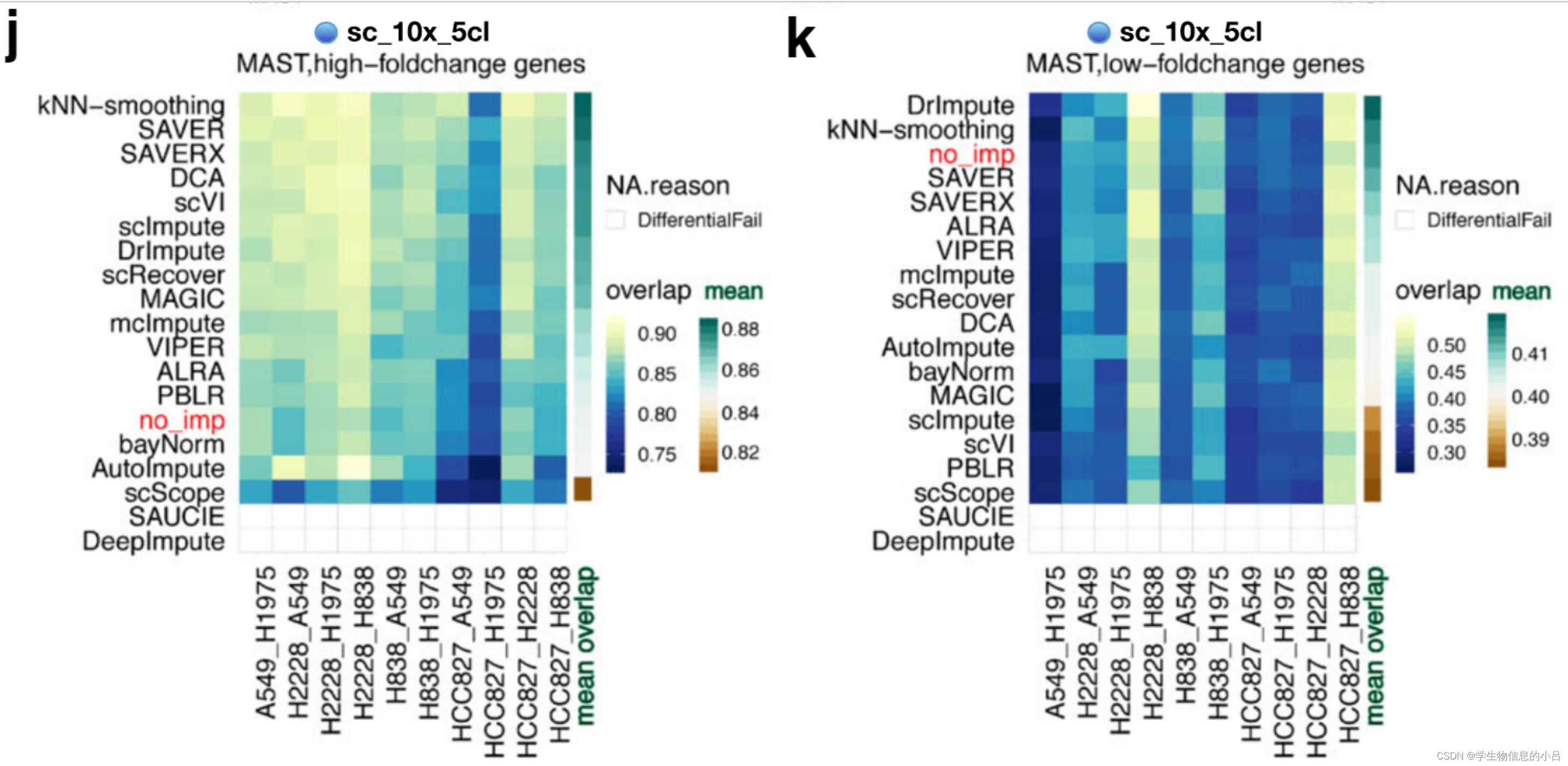
当DE的大小较大时,大多数插补方法(16种返回插补值的方法中有13种)的体细胞DEG和单细胞DEG之间的重叠程度高于无插补(图3j)。.然而,当 DE 的幅度较小时,与没有插补相比,16 种方法中只有 2 种增加了散装和单细胞 DEG 之间的重叠,这表明大多数插补方法可能已经平滑了小的差异信号 (Fig. 3k)
不同数据集有不同的信号丰度与强度,数据集合内DEG越多,imputation越容易将插补后结果与Bulk批量RNA结果重合。
大多数插补方法改进了对具有大 LFC 的 DE 基因的分析,而不是没有插补,但对具有小 LFC 的基因没有改进
结论
目前的插补方法作为一个整体,似乎对提供单个基因活性的点估计最为有效,当与各种下游分析任务相结合时,它们的效率会降低。
对于差异表达分析,有效性降低可能是由于插补后细胞方差描述不准确。对于聚类和轨迹分析,效率可能会降低,因为这两种分析试图分析细胞与细胞之间的关系,而不是单个基因。细胞聚类和轨迹分析通常通过将每个细胞的高维表达向量嵌入到一个相对低维的空间来进行。低维空间中的每个维度都包含来自许多基因的信息,这通过稀释技术噪声(如由于技术变化而观察到的零点)来增加信噪比,即使没有插补。
因此,插补对细胞间关系的恢复影响较小。相比之下,单个基因的测量包含高水平的技术噪声,通过从其他基因或细胞中借用信息进行插补,可以大大减少技术噪声。因此,插补可能更有助于分析单个基因,而不是细胞与细胞之间的关系。
未来有待研究的一个公开问题是,插补对各种下游分析任务的改进是否已经达到上限,如果没有,如何设计新的插补方法,以进一步改进考虑到细胞变异性的细胞间关系或差异表达的分析。
最新文章
- HDU 5113 dfs剪枝
- Validform:一行代码搞定整站的表单验证!
- Saltstack系列2:Saltstack远程执行命令
- HBase优化
- C++的类成员和类成员函数指针
- C语言经典算法100例(一)
- 微软职位内部推荐-ATG Engineer II
- C语言预处理运算符
- AngularJs学习笔记6——四大特性之依赖注入
- 深入Redux架构
- Cookies, Claims and Authentication in ASP.NET Core(转载)
- Tomcat实现反向代理
- [Swift]LeetCode402. 移掉K位数字 | Remove K Digits
- C#与SQL Server数据库连接
- __add__,关于运算符重载(用户权限)
- CSS实现16:9等比例盒子
- HashSet的底层
- $.fn.extend() 问:我来这个世上到底是干嘛的?
- Software-Defined Networking之搬砖的故事
- 在CentOS 6 的cron/crontab中使用wine运行exe程序